Derived from 547 genomes. For the derivation pipeline and the PASS / WARN / FAIL verdict model, see the methods page for qualibact-v1.0.
Applied to the full All-The-Bacteria dataset, these thresholds place 506 genomes at PASS, 51 at WARN, and 40 at FAIL (597 assessed in total). The per-tier genome lists can be downloaded below in .csv.gz format; the FAIL list also records the reason each assembly was rejected.
This table summarises the distribution of each metric, including standard deviation, mean, median, and percentiles.
A combined summary table across all species is available on the summary page.
| Metric | Distribution | n | Mean | SD | Min | Q1 | Median | Q3 | Max |
|---|---|---|---|---|---|---|---|---|---|
| N50 | non-normal | 547 | 235,963 | 91,611 | 38,120 | 188,000 | 237,494 | 263,984 | 505,426 |
| no_of_contigs | non-normal | 547 | 63.69 | 29.03 | 32 | 46.5 | 55 | 71 | 256 |
| longest | non-normal | 547 | 553,217 | 174,221 | 134,824 | 455,544 | 560,223 | 585,009 | 1,171,403 |
| GC_Content | non-normal | 547 | 38.2 | 0.03 | 38.01 | 38.18 | 38.2 | 38.22 | 38.32 |
| Completeness_Specific | non-normal | 547 | 100 | 0 | 99.99 | 100 | 100 | 100 | 100 |
| Contamination | non-normal | 547 | 0.26 | 0.16 | 0.04 | 0.16 | 0.19 | 0.29 | 1.25 |
| Total_Coding_Sequences | non-normal | 547 | 3,174 | 92.59 | 2,932 | 3,100 | 3,158 | 3,219 | 3,533 |
| Genome_Size | non-normal | 547 | 3,509,161 | 84,635 | 3,296,115 | 3,444,657 | 3,486,641 | 3,542,475 | 3,853,591 |
Full statistics including KS test vs RefSeq and Wasserstein distance are in the downloadable summary.csv.
Derived from 547 genomes
Both Fail and Warn bands shown as the published rounded values — easier to cite and consistent across the species page, CSV downloads, and downstream QC tools.
| Metric | Fail below | Warn below | Warn above | Fail above |
|---|---|---|---|---|
| Genome_Size | 3,300,000 | 3,300,000 | 3,800,000 | 3,900,000 |
| GC_Content | 38 | 38.1 | 38.3 | 38.3 |
| Total_Coding_Sequences | 2,900 | 3,000 | 3,500 | 3,600 |
| Completeness_Specific | 99 | 99 | - | - |
| Contamination | - | - | 1 | 2 |
| N50 | 46,000 | 76,000 | - | - |
| no_of_contigs | - | - | 170 | 190 |
| longest | - | - | - | - |
How to read this: a value between the two warn columns is typical for this species and passes QC. A value between a warn column and the corresponding fail column is borderline — worth a manual look but not an outright failure. A value outside the fail columns is unusual enough to fail QC.
The published rounded thresholds (the values in the table above) were applied to the full AllTheBacteria-2024-08 set for this species. Each row carries the per-metric verdict and, where applicable, the reason a genome was demoted to WARN or FAIL. Files are gzipped CSV.
This plot shows the relationship between the number of coding sequences (CDS) and genome size — how the number of genes scales with assembly length. The relationship should be roughly linear: as genome size increases, the number of coding sequences should rise proportionally. A secondary trend line or non-linear behaviour can indicate either bona fide sub-populations within the retained genomes (e.g. distinct sub-clades) or residual contamination that survived filtering.
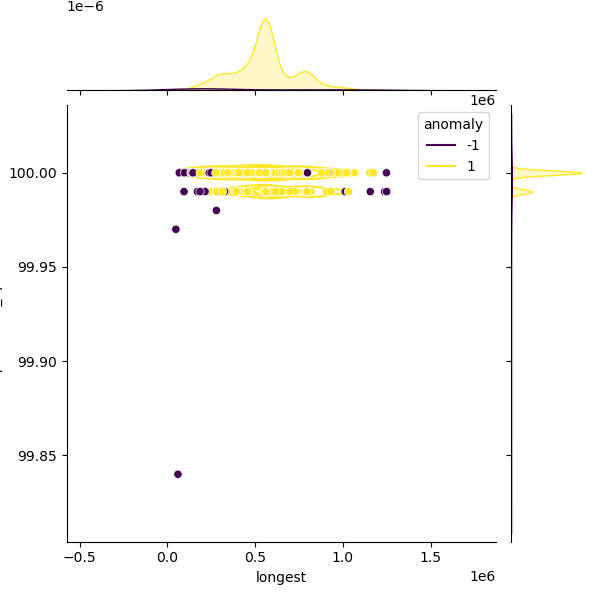
These plots show genomes before and after filtering to highlight the outliers removed:
The filtered distribution shown here may not exactly match the published thresholds because additional rounding and curator adjustments are applied on top.
{kind=link}
{kind=link}
{kind=link}